Basic Principles of Risk Management for Medical Device Design

Medical devices developed for human application are used for diagnostic or treatment purposes. They may either be an instrument, an apparatus or a material. Moreover, these devices can be used for daily patient care as well as for medical scientific purposes. Researchers in charge to develop new medical devices are faced with the complex task of making a medical device safe for human use. This implies that the device should be safe and effective. Risk management involves the identification, understand, control, and prevent failures that can result in hazards when people use medical devices. Risk Analysis plays a key role in the development of medical devices design. Risk analysis, or hazard analysis, is a structured tool for the evaluation of potential problems which could be encountered in connection with the use of taking a drug, or using a medical device. Manufacturers are expected to identify possible hazards associated with the design in both normal and fault conditions. If any risk is judged unacceptable, it should be reduced to acceptable levels by appropriate means.
The purpose of this paper is to describe the importance of Risk Analysis, Risk Management Process, Application of Risk Management tools, and the benefit of the Risk Management Analysis. The goal is to minimize use-related hazards, assure that intended users are able to use medical devices safely and effectively throughout the product life cycle, and to facilitate review of new device submissions and design control documentation.
1. Introduction
The globalization of the medical device marketplace, combined with the growth of medical device usage, has led to a significant increase the complex task of making a medical device safe for human use activity among device manufacturers. Risk Management has become an important competitive tool to gain access to foreign markets. As clinicians, patients, regulators, and litigators become more sensitive to safety issues related to human factors, the importance of appropriate translation and safety controls will increase. Risk management is necessary to ensure device usability, safety, and regulatory compliance.
In some cases, critical human factors and risk management decisions are made that depend on specific language in the user interface or labeling. For instance, hazardous situations can arise based on improper interpretation of date/time information or units of measurement displayed. Mitigation of such risks is typically a key focus during initial device development for the initial locale (e.g., the United States); however, the impact of translation and localization on these items is often not as carefully identified, controlled, verified, and validated.
FDA's quality system regulation is intended to give manufacturers "the flexibility to determine the controls that are necessary to be commensurate with risk."FDA sees risk analysis as an essential requirement of the regulation but gives little guidance on specific risk analysis approaches and procedures such as fault tree analysis (FTA) or failure mode and effects analysis (FMEA). As medical device companies review and update their approaches to risk analysis, they may find value in what other industries including chemical, aerospace, and defense have learned about using it to reduce risk. Companies can manage and reduce risk more effectively by including risk thinking as early as possible in device or process development and revisiting those issues systematically throughout the development process.
2. Overview of Risk Management
Risk management involves the identification, understand, control, and prevent failures that can result in hazards when people use medical devices. Manufacturers are expected to identify possible hazards associated with the design in both normal and fault conditions. The risks associated with the hazards, including those resulting from user error, should be calculated in both normal and fault conditions. If any risk is judged unacceptable, it should be reduced to acceptable levels by appropriate means.
2.1 Why should we perform Risk Management?
An overall risk management process involves the essential steps in Figure 1. In order to manage risk, hazards must first be identified. By evaluating the potential consequences of hazards and their likelihood, a measure of risk can be estimated. This value is compared to the company's risk-acceptability criteria and, if it is too high, the risk needs to be mitigated.

Figure 1. Risk Management Flow Chart
Because risk cannot be completely eliminated, the risk that remains must be managed. The following steps can be used in a risk management program
2.2 Risk Control
Process through which decisions are reached and protective measures are implemented for reducing or maintaining risk within the specified level.
2.3 Risk control and monitoring activities
Actions intended to eliminate or reduce each risk to meet the previously determined risk acceptability criteria.
One or more risk control measures may be incorporated.
Risk controls may begin as early as design input and continue over the medical device life time.
Some regulatory schemes prescribe a fixed hierarchy of risk controls that should be examined in the following order:
Throughout the life-cycle of the device the manufacturer monitors whether the risks continue to remain acceptable and whether any new hazards or risks are discovered.
An effective and well defined Quality Management System is key Information typically obtained from the quality management system, for example, production, complaints, customer feedback, should be used as part of this monitoring.
2.4 Risk Control Measures
Protective measures, e.g. default operating modes
Information for safety, e.g., warnings in labeling
Many measures require intervention
2.5 Safety Risk Zone
Identifies the residual safety risk for each hazard in order to make a determination of its acceptability.
Hazards that fall into Safety Risk Zone 1 (R=1) are considered generally acceptable or minimal and require no further analysis. Additional mitigations are optional for hazards that fall into Safety Risk Zone 1.
Hazards that fall into Safety Risk Zone 2 (R=2) are considered conditionally acceptable but require analysis and mitigation. These are considered acceptable only when adequate mitigations are identified.
Hazards that fall into Safety Risk Zone 3 (R=3) are generally unacceptable. If hazards in Safety Risk Zone 3 cannot be further mitigated to the point of falling into an acceptable Risk Zone (R=1 or 2), a formal risk/ benefit analysis shall be performed and documented.
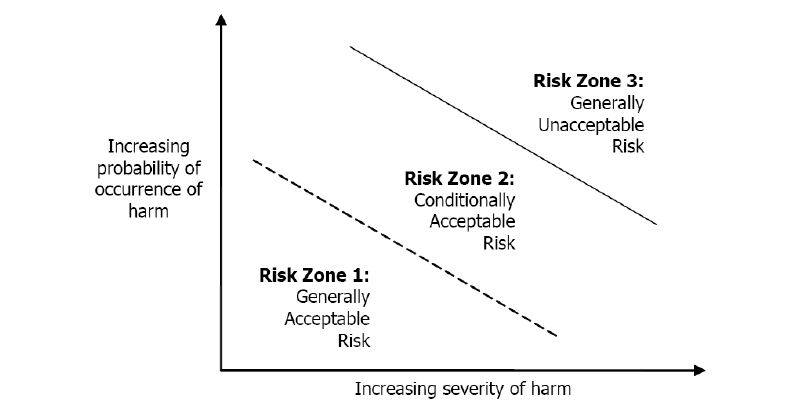
Figure 2. Safety Risk Zone
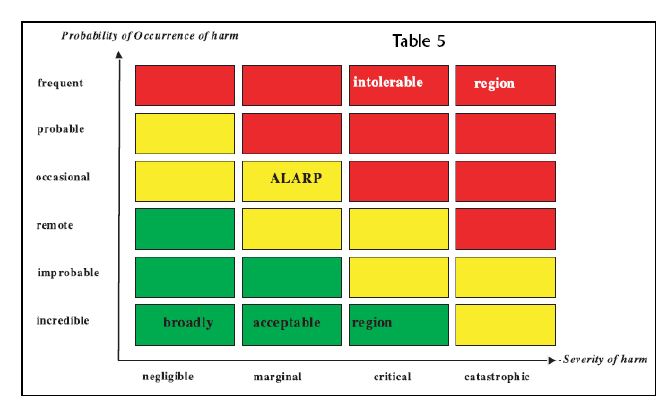


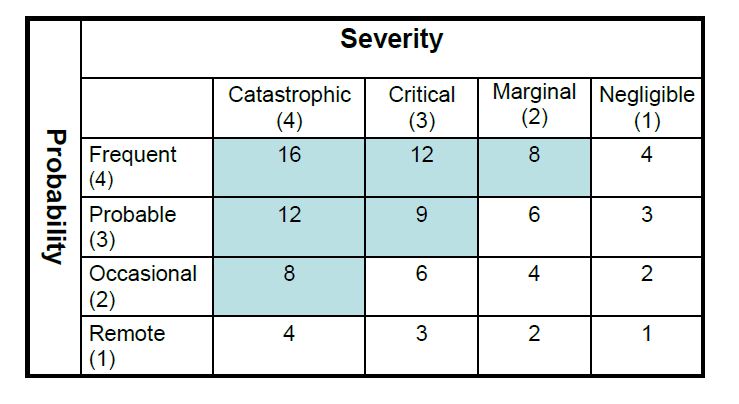
Figure 3. Risk Assessment Matrix
2.6 Mitigation
Mitigation means: What are you going to do about the situation?
1st Line of Defense Avoid or eliminate failure causes
2nd Line of Defense Identify or detect the failure earlier
3rd Line of Defense Reduce the impacts/consequences of failure
3. Hazard Analysis
Even before a final design has been developed, a preliminary hazard analysis can be conducted to establish the baseline hazards associated with a device. In essence, the analysis consists of listing the major components and operating requirements of the device and evaluating their potential hazards. The components and operating requirements could include raw materials and wastes, hardware, monitoring and control systems, human-device interfaces, services, and the operating environment.
Some potential hazards that may need to be evaluated include toxicity, flammability, and reactivity of raw materials and wastes; sensitivity to environmental factors such as temperature and humidity; mechanical or electronic hazards; and human factors associated with the operator-device interface. The patient-device interface can also be hazardous because of unsafe or ineffective delivery of energy, administration of drugs, or control of life-sustaining functions. Also, incorrect information could lead to a misdiagnosis or wrong treatment or therapy being ordered.
When conducting a preliminary hazard analysis, use a what-if or brainstorming approach to identify possible failures, evaluate potential consequences, and develop risk management strategies. These strategies lead to an improved, lower-cost design. Generally, failure scenarios can be prioritized by the severity of each hazard.
At this stage, there is often insufficient detail to evaluate hazard likelihood accurately. However, comparisons may be made with similar devices and their histories in the medical device reports. An evaluation revealing severe hazard potential may prompt a radical change in the conceptual design. The goal is to eliminate all high-severity hazards and reduce as many medium- and low-severity hazards as possible. There is considerable flexibility at this early design stage. Major changes can make the device inherently safer at minimal cost. For example, if use of a chemical was determined to be a significant hazard, other less-toxic chemicals or a diluted form of the original chemical might be a reasonable mitigating measure.
During prototype development, more detailed hazard and risk analysis can be performed. At this stage of design, process and mechanical drawings are available, and the basic process operations have been defined. The device and its operation can be reviewed by a number of analysis techniques, including top-down and bottom-up approaches. A hazard and operability (HAZOP) study is a bottom-up approach ideal for new or complex designs involving a number of processing steps. A HAZOP is conducted on individual steps, each of which has design intent.
If the deviation defined by the combination of a design parameter and guide word (e.g., more flow or less flow) can result in a hazard, potential causes and any existing controls are identified. The risk level can be evaluated using a risk matrix in which consequence and frequency ranges have been established according to a company's internal risk-acceptability criteria (Figure 2).
When a device contains many mechanical components, an FMEA should be considered. However, an FMEA is time-consuming and is generally applied only to Class III devices or to the safety critical portions of devices. For those devices that contain many electrical components, an FMEA is also a desirable methodology. This is another bottom-up approach that focuses on a particular component of a medical device and explores the various failure modes that can occur. For each failure mode that results in an undesirable consequence, potential causes and existing controls are evaluated, and the level of risk can be determined by using a risk matrix.
An FTA is an effective top-down approach. The team starts with the undesired consequence or top event and identifies the initiating and contributing events that must occur to produce it. These events are combined using logic gates. A logic gate is the point at which two or more independent events are combined in order to produce a higher-level event. The logic gate determines whether the sub event probabilities or frequencies should be multiplied, for an and gate, or added, for an or gate. If all events under a gate are necessary for the higher event to occur, an and gate is used. If each of the events is sufficient to produce the higher event on its own, an or gate is used. Both mechanical failures and human errors can readily be included in a fault tree. An example of a partial fault tree for a pacemaker is shown in Figure 4.
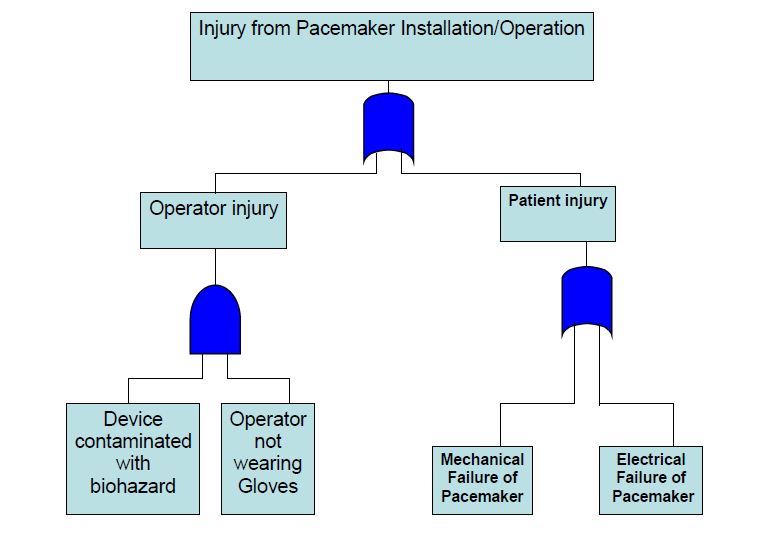
Figure 4. A Partial Fault Tree Analysis for a Pacemaker
The top event is an injury resulting from installation or operation of the device. Below the top event are two sub events labeled operator injury and patient injury. Since either could produce the top event, they are combined using an or gate. Under the operator injury branch, one potential scenario has been identified that involves having the device contaminated with a biohazard such as blood (the initiating event) and the operator not wearing gloves (contributing event). Since both the initiating and contributing events must occur for an injury to take place, these events are combined using an and gate.
If failure rates for each event on a fault tree are available or can be estimated from generic data, the top-event frequency can be calculated and compared to a company's internal risk-acceptability criteria. A fault tree is a powerful risk-analysis tool, but its greatest limitation is the availability of relevant failure data. Therefore, fault trees are generally best used to compare risks of various alternatives. The greatest benefit of a fault tree is that events that contribute most frequently to the top event can readily be identified, and mitigating measures can be focused on reducing the frequency of these events.
4. Procedure Analysis
Although HAZOP, FMEA, and FTA allow evaluation of human errors in design, operation, and maintenance of medical devices, it is often desirable to conduct a separate analysis focused on procedures. Typically, a what-if approach is used for this type of analysis. Procedures are grouped into process steps similar to those study sections used with HAZOP. Each process step is evaluated to determine if an undesirable consequence could result from incorrect procedures.
Checklists are the simplest tools for conducting design reviews but are generally not sufficient. The true benefit of checklists is to support the other techniques described previously. For example, a checklist of potential hazards identified in previous reviews or from incidents associated with similar devices would be useful during a design review. After completion of the review, the checklist can be examined to ensure that the study evaluated all previously identified potential hazards. For example, during a HAZOP, possible human errors are evaluated; however, as a final check, a human-factors checklist is often used.
The risk analysis should include any risks associated with the manufacture and delivery of the device to its intended location. For devices that involve solutions or components that can be degraded by environmental factors (e.g., heat, humidity, cold, or light), storage and transportation methods need to be reviewed. Identified problems could lead to changes in packaging or warnings on storage or packaging containers.
It is important that any changes made during the design process be reviewed to ensure that safety hazards are not being introduced into the design. Small changes are generally reviewed using a what-if approach, whereas larger changes may require a HAZOP or FMEA.
A final design or prestart-up review should be conducted before starting production. Extensive checklists ensure that all design specifications have been met and all previous design review recommendations have been addressed. The final design review should also include a physical inspection of the device in its intended workspace (e.g., laboratory, hospital, doctor's office) to identify any issues not readily apparent from looking at drawings, such as location of vents and drains, accessibility for maintenance, pinch points, and sharp edges. A punchlist (findings or observations developed during a safety or design review) of final action items is typically generated and prioritized into items that need to be completed prior to start of production and others that can be incorporated into the next model.
Software used to control or monitor a medical device also needs to be reviewed. Software can be grouped into its primary functions (e.g., start-up, treatment, diagnostics, and maintenance) just as procedures can be grouped into process steps. Three generic sub functions are evaluated for each primary function:
Software errors can produce unexpected consequences, particularly those that involve corrupt data or false alarms. It is important to have a means of detecting software errors or a means to detect the effects of software errors on a device. For example, a software error resulting in a failure of the alarm notification system would disable all alarm systems. Separate redundant alarms or interlocks on critical aspects of a device need to be considered.
5. ISO 14971:2007
It is the specified standard for risk management used to demonstrate compliance with the Risk Management requirements of the Medical Devices Directive (MDD).
The standard addresses risk management to patient, operator, other parties, external equipment and/or the environment. Risk Management Process ISO 14971 requires the manufacturer to establish, document and maintain a risk management process for:
The risk management process does not end with the design and manufacturing process but also includes applicable sterilization, packaging, labeling, storage, handling/ transport, distribution and market surveillance. The manufacturer shall apply risk management from the initial conception until the ultimate decommissioning and disposal of the product. Therefore, the gathering of post- production information is a required part of the process.
The latest version of ISO 14971:2007 (“Medical devices – Application of risk management to medical devices”) was approved on 5 December 2006 by the Association for the Advancement of Medical Instrumentation (AAMI) and on 1 February 2007 by the American National Standards Institute (ANSI). Finally published in May 2007 as ANSI/AAMI/ISO 14971:2007
6. Conclusion
All of the techniques described above have been successfully used in design reviews of medical devices. FTA is being used by pacemaker manufacturers based on FDA guidance for software aspects of 510(k) notification submissions for medical devices. Other computer-controlled medical devices will also need to be reviewed using FTA as a primary risk analysis tool.
For mechanical devices that are used away from the patient, such as plasma and blood viral inactivation devices, as well as devices for preparing intravenous solutions, an FMEA is a reasonable choice. However, for associated activities such as preparation of disposables, which are manual operations, a what-if approach is preferred.
The key to successful risk management in medical device design is to start early. As soon as conceptual designs are available, the risk management process can begin. A preliminary hazard analysis can be useful in selecting the concept with the highest level of inherent safety. Later, as the design is developed, design reviews at key points in the development process will allow changes to be made without significantly affecting the project schedule. The further along in the design process that changes are identified, the fewer choices are available to mitigate hazards without significant schedule implications.
Generally, risk management activities will identify opportunities to improve device performance. The benefits of conducting risk analysis during medical device design can be significant and can be used to offset some or all of the cost of implementing risk-mitigating measures. There is always a trade-off in how to manage risk. Hardware or software controls are generally viewed as more effective since they are more reliable than humans. However, since there is need for human interaction in the operation of all medical devices, the element of risk needs to be adequately evaluated. Minimizing the level of routine human intervention will reduce risk and improve efficiency. Such risk reduction must be weighed against the cost of automating tasks that can be performed by individuals.
References